MSeqDR Quick-Mitome Tutorial
Lishuang Shen (lishen@chla.usc.edu )
A Phenotype-Guided WES/WGS Variant Interpretation Server for Mitochondrial Diseases
1. Step 1. Upload variant VCF (v4+, the sample column is required) and pedigree file:
1.1. Add VCF file:
1.1.1. Drag & Drop VCF file (*.vcf or *.vcf.gz) to the uploadbox to upload.
1.1.2. Click the blue upload button or icon to upload
1.1.3. After VCF file uploading, click Refresh link to see the files
1.1.4. Use VCF format version 4.0 or above, the sample genotype column is required. Chromosomes must be named as '1, 2, .. X, Y', or 'chr1, chr2, ... chrX, chrY'.
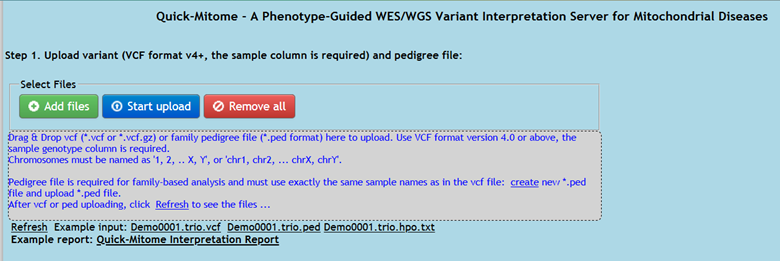
Figure 1. VCF file and pedigree file upload box.

Figure 2. Drag-and-drop a VCF file into upload box.
1.2. Pedigree file preparation
1.2.1. If you already have an existing family pedigree file (*.ped format) , Drag-and-drop it to the uploadbox to upload, same as for vcf upload. After uploading, click Refresh link to see the file.
1.2.2. If you need to create a new pedigree file:
1.2.2.1. Click the "Check vcf" button, the sample names are displayed in "Sample in vcf" box.
1.2.2.2. Click the "Create PED" button to go the tool "MSeqDR Pedigree File Generator"
1.2.2.3. In the tool page, use the Proband name as Family name, then the Proband, Father, Mother and sibling information as available in the vcf file. Setup gender and affected status information
1.2.2.4. Click "Create PED file" button, review the PED file in the textarea, and save save the pedigree PED file for the family. One copy is automatically added to server .
1.2.3. After PED file creation or uploading, click the Refresh link to see the file in "Pedigree files available".
1.2.4. Pedigree file is required for family-based analysis and must use exactly the same sample names as in the vcf file (1.2.2.1).
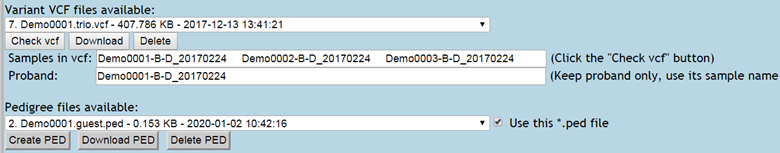
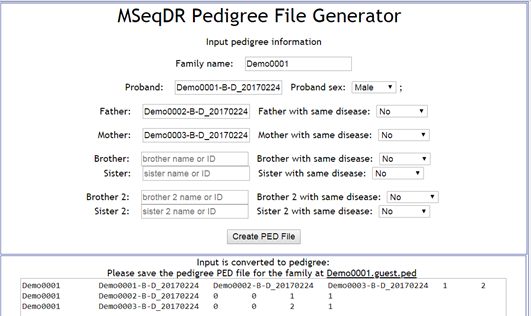

Figure 3. Pedigree file preparation by A. extracting sample information from VCF file (top), B. Input the family and sample information in "MSeqDR Pedigree File Generator" (middle), and C. Result pedigree files shown after clicking the "Refresh" link (bottom).
2. Step 2. Input clinical symptoms and diagnosis description and double click "HPO Annotator" button (Figure 4)
2.1. One type of input is an existing HPO term list to be pasted into the "HPO ID" box, separated by commas.
2.2. Another type of input is free text clinical description.
2.2.1. Copy-pasted the free text clinical description into the text area, one term per line
2.2.2. Click the "HPO Annotator" button, then the terms are mapped to HPO terms, details are shown at the page bottom for review.
2.2.3. Candidate terms are extracted and ranked by semantic similarity to HPO term names, synonyms, and definitions.
2.2.4. The best HPO match is automatically picked and selected with checkbox. Users can review other HPO matches and use checkbox to override the automatic picks.
2.2.5. All the picked HPO terms are automatically converted into Quick-Mitome input, saved into the "HPO ID" box.
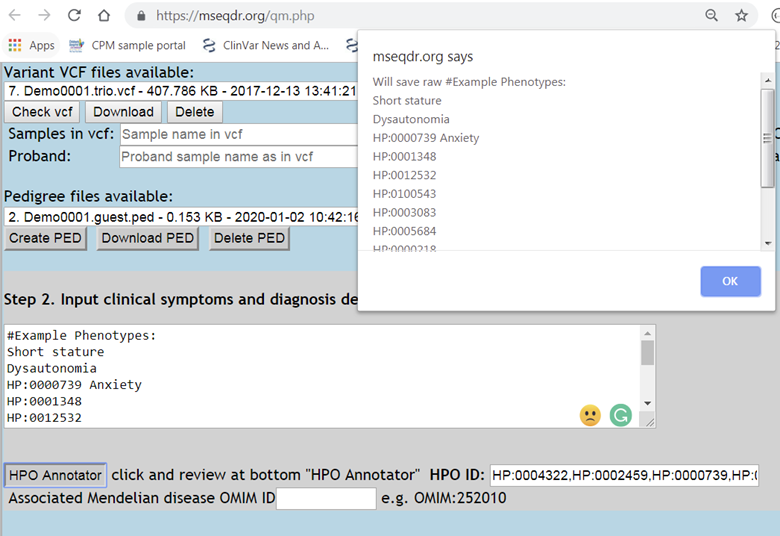
Figure 4. Clinical description (free text) mapping to HPO IDs..
3. Step 3. Run annotation and interpretation using the above selected VCF, pedigree (if multiple sample vcf), and HPO term IDs (Figure 5)
3.1. Select the VCF from VCF file drop-down list,
3.2. Select a matching pedigree file from pedigree file drop-down list if this is a multiple sample vcf
3.3. Designate the proband name in "Proband name" box. It must be exact the same sample name as in vcf.
3.4. HPO term IDs are input into the "HPO ID" box, separated by commas
3.5. Over 10 MSeqDR Tools are designed to run automatically right now, or will be called from the Quick-Mitome Interpretation Report tool. No setting change is needed.
3.6. Although not required, it is strongly recommended to fill in your email to receive result notice and report links for permanent record.
3.7. Click the "Run Annotation and Interpretation" button.
3.8. Do not re-submit or refresh, must leave this page to run till it is completed. The run may need about 10 minutes for input with 1500 variants.
3.9. Results will be displayed under "Step 4. Check Annotation and Interpretation Report"
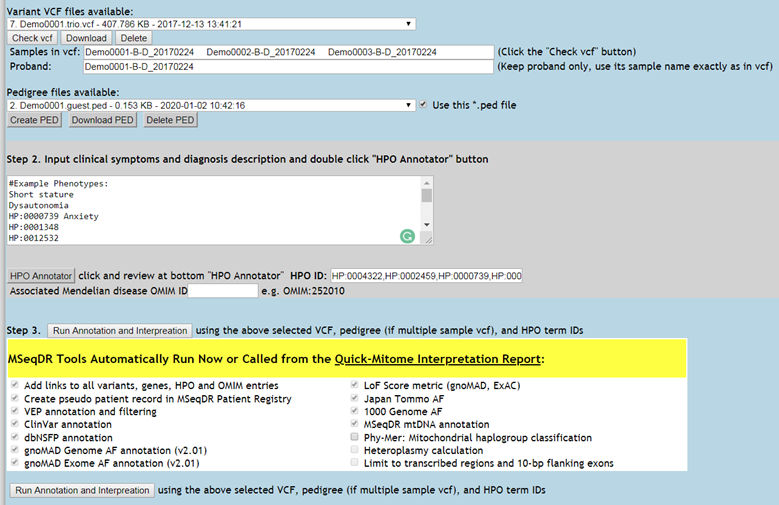
Figure 5. Set up annotation with the VCF, pedigree, HPO ID, and (optional) email address.
4. Step 4. Check Annotation and Interpretation Report (Figure 6)
4.1. The run summary, links to report, and timing are displayed here. And emailed to your email address if choose to.
4.2. Each run is renamed with a 50-character randomized ID to protect data security, i.e. CRJVJVNZ6RG3CTZN8CGJRMZA9HG9NERFNS65LHTR7RAG8GJFYA . This ID must be saved for retrieving your analysis report in future. Valid email will receive automatic copy of the information.
4.3. The top part is the Your Quick-Mitome interpretation report, and Mito-disease variant labeling report to call the deep-learning
mitochondrial-disease variant classifier (Figure 6, top part).
4.4. The bottom part includes the Exomiser analysis log, the combined HTML report, the variant and gene report per inheritance mode (AD, AR, XD, XR, or Mitochondrial). All variant, gene, HPO and OMIM ID entry in the HTML are hyperlinked to MSeqDR and external resources (Figure 6 bottom part and Figure 7).
4.5. Click on the link "Your Mito-disease
variant labeling report". It will generate the Mito-disease variant classifier input of features. The variants are filtered with 2 filters: Filtering criteria 1 is allele frequency: gnomad_genome_AF_POPMAX<=0.05 AND gnomad_genome_AF
<=0.02, and filtering criteria 2 is variant consequence: Exclude nocoding variants of 'intron_variant', 'downstream_gene_variant', and 'upstream_gene_variant'.
4.6.
Then click the link "Make deep-learning prediction of variant pathogenicity, plus mitochondrial-disease causing probability". Result is shown as in Figure 8.
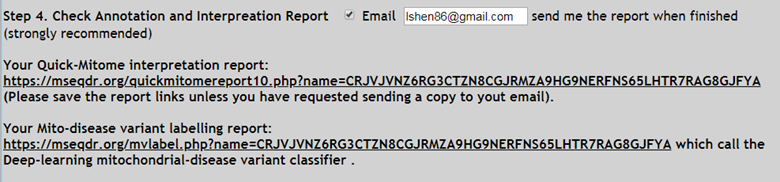
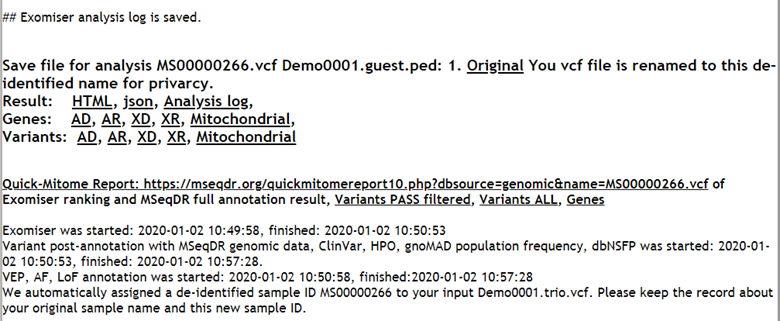
Figure 6. Quick-Mitome annotation report (top), and the Exomiser run information (bottom).

Figure 7. Quick-Mitome report part 1.
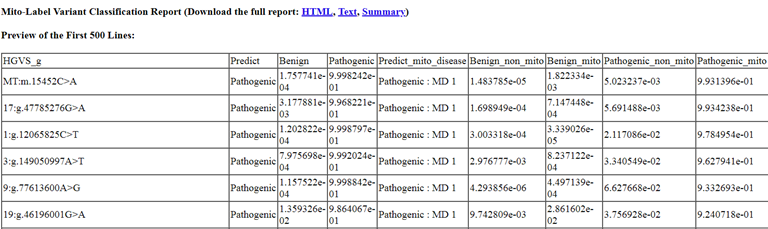
Figure 8. Quick-Mitome report part 2- p-values of variant pathogenicity, plus mitochondrial-disease causing probability.
5. Example Input, Annotation, and Interpretation Report
5.1. Example VCF file (Trio): Demo0001.trio.vcf
5.2. Example Pedigree file (Trio): Demo0001.trio.ped
5.3. Example Phenotype (HPO): Demo0001.trio.hpo.txt HP:0002817,HP:0040070,HP:0009810,HP:0000739,HP:0001348,HP:0012532,HP:0100543, HP:0003083,HP:0005684,HP:0002459,HP:0000218,HP:0002828,HP:0004322
5.4. Example Quick-Mitome interpretation report (Figure 7): CRJVJVNZ6RG3CTZN8CGJRMZA9HG9NERFNS65LHTR7RAG8GJFYA
5.5. Example Mito-disease variant labeling report (Figure 8): CRJVJVNZ6RG3CTZN8CGJRMZA9HG9NERFNS65LHTR7RAG8GJFYA , which calls in realtime the Deep-learning mitochondrial-disease variant classifier.